
DNA Sequencing
How it works
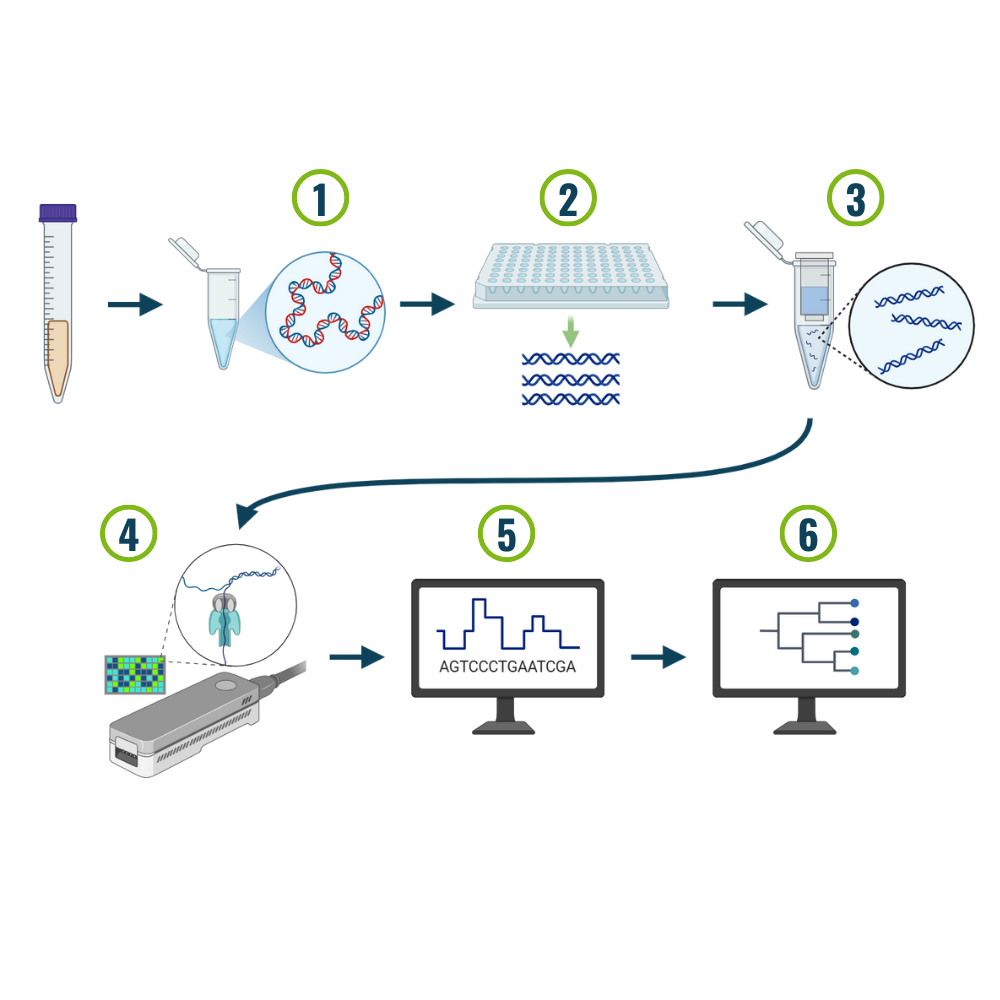
1. DNA Extraction: The cells in the sample are lysed (broken apart) and the pure DNA is extracted. In our workflow cells are lysed using a mechanical method called bead beating, in which tiny glass beads are mixed with the sample and then the mixture is shaken aggressively by a specialized machine. Cells can also be lysed using chemicals, heat, or freeze-thaw methods.
2. PCR Amplification: We use 2 different genes to identify the microbes in each sample. The 16S gene is used to identify the bacteria present, and the ITS gene is used to identify the fungi / oomycetes. Using PCR (polymerase chain reaction) we are able to “photocopy” or amplify our genes of interest so that we can sequence them specifically.
3. PCR Purification: The final PCR mixture is then purified to remove all of the proteins and extra little bits of DNA that are byproducts of the PCR reaction. These contaminants in the PCR reaction can clog up the pores on the sequencer, causing the sequencing run to fail or perform poorly.
4. Sequencing: Long read sequencing is then conducted using an Oxford Nanopore Sequencer. This type of sequencing method can be used to obtain the full 16S or ITS sequences (approximately 1,600 and 800 base pairs long), which are much longer than older technologies like Illumina sequencing. This longer sequence gives us more information to correctly identify each species (think of it like identifying a book from either a paragraph or a sentence).
5. Base Calling: Nanopore sequencing works by measuring the change in electrical current along a membrane as each base passes through a pore. Base calling is the process of taking those electrical read outs and turning them into a DNA sequence.
6. Species Identification: The DNA sequences are compared against a database of previously identified 16S/ITS genes to determine which microbes are present in each sample. Based on the number of 16S or ITS reads we are able to determine the relative abundance of each organism within each sample.